
By Robert J. Charlson[1], James E. Lovelock[2], Meinrat O. Andreae[3] & Stephen G. Warren[1]. Published in Nature Vol. 326 No. 6114. pp. 655-661, 16 April 1987.
[1] Department of Atmospheric Sciences AK-40, University of Washington, Seattle, Washington 98195, USA.
[2] Coombe Mill Experimental Station. Launceston. Cornwall PL15 9RY, UK.
[3] Department of Oceanography, Florida State University, Tallahassee, Florida 32306, USA.
Abstract: The major source of cloud-condensation nuclei (CCN) over the oceans appears to he dimethylsulphide, which is produced by planktonic algae in sea water and oxidizes in the atmosphere to form a sulphate aerosol. Because the reflectance (albedo) of clouds (and thus the Earth’s radiation budget) is sensitive to CCN density, biological regulation of the climate is possible through the effects of temperature and sunlight on phytoplankton population and dimethylsulphide production. To counteract the warming due to doubling of atmospheric CO2, an approximate doubling of CCN would he needed.
Climatic influences of the biota are usually thought of in connection with biological release and uptake of CO2 and CH4 and the effect of these gases on the infrared radiative properties of the atmosphere1. However, the atmospheric aerosol also participates in the radiation balance, and Shaw2 has proposed that the aerosol produced by the atmospheric oxidation of sulphur gases from the biota may also affect climate. So far the physical and biological aspects of this intriguing hypothesis have not been quantified, but three recent discoveries may make this possible for the remote marine atmosphere.
- Most species of phytoplankton, ubiquitous in the oceans, excrete dimethylsulphide (DMS) which escapes to the air where it reacts to form a sulphate and methane sulphonate (MSA) aerosol.
- This non-sea-salt sulphate ( NSS-SO42- ) aerosol is found everywhere in the marine atmospheric boundary layer.
- Aerosol particles which act as cloud-condensation nuclei (CCN) in the marine atmosphere are principally, perhaps almost exclusively, these same NSS-SO42- particles.
In this paper we show that emission of DMS from phytoplankton is sufficient to justify its consideration as the gaseous precursor of CCN in the remote and unpolluted marine atmosphere. We re-examine the physical role of the sulphate aerosol in atmospheric radiative transfer, particularly in clouds, which are responsible for most of the Earth’s albedo, and estimate the sensitivity of the Earth’s temperature to changes in the abundance of CCN. Finally we examine the geophysiology 3 of the system comprising phytoplankton, DMS, CCN and clouds, as a putative planetary thermostat.
Production of sulphur-containing gases
Assuming that sulphuric acid and sulphate aerosols are indeed the only significant contributors to CCN over the oceans (as will be concluded below), we have to consider what processes are responsible for the production of the volatile sulphur compounds that are the precursors of sulphuric acid in the atmosphere. In this discussion, we shall ignore the perturbations of the atmospheric sulphur cycle by manmade fluxes of SO2 (mostly from burning of coal and oil) and shall consider only the natural fluxes, which currently represent about 50% of the total flux of gaseous sulphur to the atmosphere, and which still dominate the atmospheric sulphur cycle in the Southern Hemisphere4-6.
The only significant non-biological natural flux is the emission of SO2 and H2S by volcanoes and fumaroles. This process releases of the order of 0.4 Tmol yr-1. about 10-20% of the total natural flux of gaseous sulphur to the atmosphere4,5. The emission of sulphur by volcanoes is highly variable in space and time. Consequently, the production of sulphate aerosol by the oxidation of volcanic sulphur during the quiescent stage of a volcano is of regional importance only. Large eruptions, on the other hand, which emit enough gaseous sulphur compounds to influence wider areas, are relatively rare events. For this discussion, we shall assume that the contribution of CCN from volcanic sulphur to the global atmosphere is proportional to its contribution to the total sulphur flux, that is, 10-20% of the natural component at present.
Volatile sulphur compounds are emitted both by terrestrial and marine biota. The marine emissions are almost exclusively in the form of DMS, whereas the emissions from land are in a variety of chemical species, including H2S, DMS, methanethiol, CS2, COS and others. This difference is related to the biological processes which are responsible for the production of the sulphur volatiles. Most sulphur at the Earth’s surface is present as sulphate, which is the thermodynamically stable form of sulphur in the presence of oxygen. Sulphate is reduced by organisms through two mechanisms, ‘assimilatory’ and ‘dissimilatory’ sulphate reduction. The dissimilatory pathway is restricted to sulphate-reducing bacteria in anaerobic environments; due to physical and microbial restrictions only a small fraction of the H2S produced by this process can escape to the atmosphere7. The products of the assimilatory pathway are a variety of organosulphur compounds, the largest fraction being the amino acids cysteine and methionine, which are incorporated into proteins.
On the continents, the breakdown of organosulphur compounds during fermentative decomposition of organic matter is probably the most important mechanism for the release of sulphur volatiles. Due to the difficulty in obtaining representative data from land biomes, the magnitude of this flux is still poorly known. Estimates range from about 0.15 to 1.5 Tmol yr-1; the best estimate appears to be around 0.25 Tmol yr-1 for the total land surface, or about 2 mmol m-2 yr-1, which turns out to he a sightly smaller flux per unit area than is emitted from the oceans (about 3 ± 1.5 mmol m-2 yr-1). Preliminary data from the tropical regions suggest that there may be a net transport of biogenic sulphur from the marine to the continental atmosphere, consistent with a smaller emission flux per unit area on the continents8. Coastal wetlands play a surprisingly small role in the global sulphur cycle; at present we estimate their contribution to be about 2% of the total gaseous emissions5. This is because although the emission per unit area may be relatively high, the total area of coastal wetlands is quite small. In the marine environment, most volatile sulphur is emitted in the form of DMS which is excreted by living planktonic algae.
In contrast to microbial decomposition which produces a complex mixture of volatile sulphur species, algal metabolism yields DMS as the only volatile sulphur species. The biological function of the production of DMS is still unclear. The substance from which DMS originates, dimethylsulphonium propionate (DMSP), is important in osmoregulation in a number of phytoplankton types9 and also participates in the biochemical cycle of methionine. DMSP is also excreted by algae, and its breakdown in sea water may release additional amounts of DMS.
Even though DMS is excreted by phytoplankton, its concentration in surface sea water, and consequently its emission rate to the atmosphere, is only weakly correlated with the usual measures or phytoplankton activity, for example, chlorophyll concentration or T-uptake rate. In fact, the calculated flux of DMS from the tropical oceans, which have a low primary productivity, is essentially the same per unit area (2.2 mmol m-2 yr-1) as from the much more productive temperate oceans (2.4 mmol m-2 yr-1)5. Recent evaluations of seasonal effects10,11 suggest that the annually averaged flux from temperate regions may even be considerably lower than the value given here. Even the highly productive coastal and upwelling regions do not support much higher DMS fluxes (5.7 and 2.8 mmol m-2 yr-1), respectively, based on a large body of data from several research groups5). It appears that, independent of the rate of primary production, the warmest, most saline, and most intensely illuminated regions of the oceans have the highest rate of DMS emission to the atmosphere. This is a key fact to keep in mind when discussing possible climatic feedback mechanisms.
Two reasons can be given for this unexpected behaviour. First, the concentration of DMS in surface sea water depends not only on the rate of DMS production, but also on its rate of removal. The two dominant removal processes are ventilation to the atmosphere and the photochemical and microbial breakdown of DMS in the water column12,13. The rate of microbial removal of DMS will increase both with the density of bacteria which are able to metabolize DMS and, in a nonlinear fashion, with the concentration of DMS in sea water. The density of bacterioplankton is a function of the densities of phytoplankton and of grazing organisms. Due to these diverse and nonlinear relationships between the variables which regulate DMS production and removal, we cannot expect to find a simple, linear relationship between phytoplankton density and DMS concentration. Furthermore, the production rate of DMS shows variations over three orders of magnitude among different phytoplankton species (ref. 14 and M.O.A., unpublished data). It appears that some algal groups, such as the coccolithophorids, which are most abundant in tropical, oligotrophic waters, have the highest rate of DMS excretion per unit biomass. We conclude. therefore, that the global input of DMS into the atmosphere is much less sensitive to changes in total primary production than to variations in phytoplankton speciation. In other words, we can accommodate rather large swings in marine productivity without changing the climatic effects produced by the sulphur cycle, and on the other hand. we could essentially eliminate the marine input of sulphur to the atmosphere without significantly changing the total primary productivity, for example by replacing all coccolithophorids with diatoms. The concentration of SO42- in sea water is so large (~29 mmol kg-1) that it does not limit the emission of DMS.
We can now evaluate the role of sulphur gases in general (and DMS in particular) as sources of NSS-SO42- and of CCN in the unpolluted marine troposphere. Because sub-micrometre NSS-SO42- particles are not directly emitted by land or ocean surfaces, they are created in the atmosphere only by chemical reactions. As far as is known, the only significant gaseous sulphur precursors of NSS-SO42- of biological origin are DMS from the oceans and H2S, DMS and perhaps other sulphur species from land biota. These gases are oxidized in air, largely by OH15,16,75, to form SO42-. At the low N0x concentrations typical of the unpolluted marine troposphere, the oxidation of DMS by NO3, can be considered negligible17-18. In addition to SO2 the oxidation of DMS by OH also produces MSA. At low NOx levels, however, the fraction of MSA produced is less than 20%, as suggested both by laboratory experiments (ref. 19 and unpublished manuscript by I. Bames, V. Bastian and K. H. Becker, 1986) and by the ratio of MSA to NSS-SO42- observed on marine aerosols from remote regions20,21. MSA is mostly present as aerosol particles (M.O.A., unpublished data) and due to its high solubility is also likely to be effective as CCN.
The oxidation of DMS to DMSO by the IO radical, which has recently been proposed (unpublished manuscript by I. Barnes et al), is probably not important as a sink for DMS because the concentrations of DMSO in the atmosphere and in marine rain have been found to be very low compared to those of DMS. MSA and NSS-SO42- (ref. 22 and M.O.A., unpublished data). The present evidence thus suggests that SO2 is the major oxidation product of DMS in the unpolluted marine atmosphere. In the presence of cumulus clouds, which are abundant in the marine boundary layer, conversion of SO2 to NSS-SO42- is rapid and significantly exceeds the rate of dry deposition of SO2 (refs 18, 21).
The rates of DMS oxidation obtained from laboratory experiments, and the corresponding lifetimes, are consistent with observed concentrations, the observed flux out of the atmosphere in rain and the calculated flux into the atmosphere from the ocean surface5,17,18. Thus, the lack of alternative sources of sulphur-bearing gases and the internal consistency of mass-balance calculations support the hypothesis that these biologically derived gases are the probable main sources of NSS-SO42- in the remote marine boundary layer. It is therefore reasonable to assume that any change in atmospheric DMS concentration would cause a corresponding change in NSS-SO42- concentration and hence in the number of particles which act as CCN. This could be due to changes in the number-concentration of particles, changes of the mass of water-soluble material in existing particles, or both.
Effect on radiative properties of clouds
Clouds of liquid water droplets form only in the presence of CCN. In the unpolluted marine atmosphere, the concentration of particles capable of being CCN varies from about 30 to 200cm \ depending on aerosol content, supersaturation and meteorological conditions21. In this same environment, the total number of submicrometre particles is often about 200cm-3 such that a significant fraction of them must be CCN. Bigg24 made observations of CCN at Cape Grim. Tasmania, when the total number-concentration of particles was less than 300 cm-3 Results for relative humidities between 100.3% and 101% (values believed to be typical of marine clouds) showed that 40% to 80% of the particles in that marine setting were active CCN. Thus, changes in NSS-SO42- in marine air are expected to result in changes in the number-density of cloud droplets, contrary to the widely held belief supported by Fletcher5 that the atmosphere always has an overabundance of particles that could act as CCN. Present-day continental air, by contrast, probably is consistent with Fletcher’s generalization.
Koehler26 argued that most CCN are composed of watersoluble materials, such that the marine NSS-SO42- should qualify. It is observed that sea-salt particle concentrations at cloud height are typically not more than 1 cm-3 so that sea-salt itself cannot be the main CCN23,27,28. Water-soluble materials other than sea-salt and NSS-SO42- do exist in marine air but the data are not extensive. Nitrates probably are found on coarser particles, and hence do not contribute substantially to the number population29. Organic compounds exist in the submicrometer particles, but their mass-concentration is probably only a tenth that of NSS-SO42- (W. H. Zoller, personal communication). Their number-concentration and sources are unknown.
The idea that natural CCN in marine air consist of sulphates has been believed for decades and that they have a widespread gaseous precursor was suggested by Hobbs27. DMS was identified as the likely source of most particles in marine air by Bigg et al30. The size distribution of submicrometre NSS-SO42- as deduced from size-resolved samples31,32 is about right for activation at supersaturation between 0.1 and 1 which is appropriate for marine clouds and is comparable to the values used by Bigg24. The mass-concentration of NSS-SO42- in the remote marine troposphere is about 0.3 μg m-3 (refs 31, 33) which, if the number-mean radius is 0.07 μm (which is reasonable76) yields a total number-population of about 100 cm-3, in agreement with measured CCN populations. The concentration of NSS-SO42- in remote marine rain water is around 2-5 x 10-6 M (ref. 34), which agrees with a simple nucleation scavenging calculation with 0.2-0.5 μg m-3 of NSS-SO42- aerosol and 1 g m-3 liquid water in the cloud35.
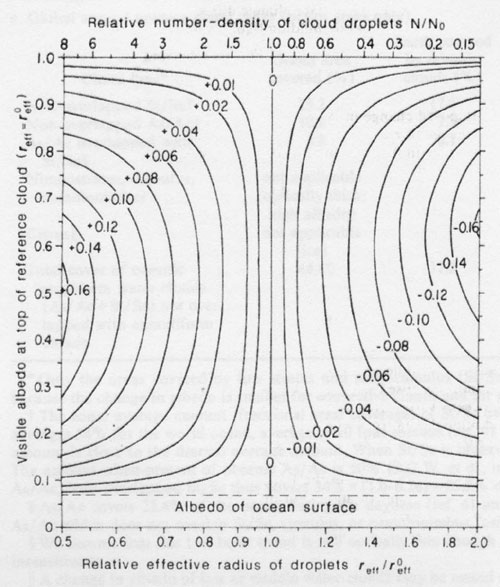
Figure 1. Change (Δa) of visible albedo (0.6-μm wavelength) at cloud top caused by changing droplet number-density N while holding vertically integrated liquid water content (liquid water path. LWP) constant. Δa is plotted as a function of the albedo of the reference cloud and the effective radius (reff the surface-area-weighted mean radius65) of the dropsize distribution relative to that of the reference cloud (reff0). The corresponding change in top-of-atmosphere albedo (needed for estimating the effect on Earth radiation budget) can be obtained approximately by multiplying these values by 0.8. as described in footnote of Table 1. The different albedos for the reference cloud (vertical axis) correspond to different values of LWP. When LWP and the shape of the dropsize distribution are held fixed, the number-density of cloud droplets N is related to reff by N/N0= (reff / reff0)-3 shown on the scale at the top of the figure. These calculations used reff0 = 8 μm, but the figure also is approximately valid for other reference clouds: the plotted values of Δa are in error by less than 20% if albedo < 0.9 and 4 <= reff0 <= 500 μm (that is, anywhere in the range of reff found in real clouds by Hegg66). The size distributions used for the calculations arc almost monodisperse, broadened just enough to average over the oscillations in the Mie-scattering quantities. However, the calculations are also valid for any realistic size distributions with the same reff as Hansen and Travis65 showed that the scattering properties of a cloud are controlled essentially by reff. wth very little influence from other moments of size distribution. These calculations assume a direct solar beam at the global average zenith angle Θ0=60°, incident on a cloud of spherical droplets of pure water, above an ocean surface. The albedo of an ocean surface under a cloud is essentially independent of wavelength and averages 0.06-0.08 (refs 67-69); these calculations assumed 0.07 (dashed horizontal line). The computation of phase function, single-scattering albedo and extinction efficiency for individual cloud droplets used the Mie program of Wiscombe70 assuming the refractive index for water is 1.332-1.09+10-8i at 0.6 μm wavelength71. The computation of radiative transfer in the cloud used the delta-Eddington approximation72. This leads to absolute errors in alhedo (for water-clouds at visible wavelengths and Θ0=60°) of 0.00 to 0.03 depending on cloud optical thickness (Fig. 8 of ref. 73), but the error in albedo differences plotted here is much smaller, generally by a factor of 10.
Other data also support the idea that NSS-SO42- particles are the main contributor to the CCN. Much of the light-scattering aerosol in marine air is volatile at elevated temperatures, evaporating in a fashion identical to sulphates with a range of compositions from H2S04 to (NH4)2SO4, as deduced from temperature-and-humidity-controlled nephelometry76,77. The droplet-nucleating property of particles in marine air is also heatlabile, with CCN disappearing at T > 300°C (ref. 36). Finally, the turnover time of CCN from purely physical data in the atmosphere has been deduced to be of the order of one day37, which is the same as results from an estimate for turnover time of NSS-SO42-based on mass-concentration, rainfall concentration and rainfall amount, as follows. If we take an NSS-SO42- flux, F, of 0.3 g m-2yr-1 as representative of remote marine sites34, 0.3 μg m-3 as the typical concentration, C, of NSS-SO42- aerosol, and 3,000 m as its scale height, H, the turnover time r=HC/F ~ 1 day.
We now consider the potential effects of NSS-SO42- variations on cloud properties. Changing the size distribution or concentration of the CCN causes the size distribution of cloud droplets to change38, which could affect the coalescence and rain production process and possibly the time-averaged cloud cover. However, the effect which is well established (and which we think to be the most significant effect) is that changes in the size distribution of droplets would change the reflectance (albedo) of clouds. This step of the proposed climatic feedback loop is at present more readily quantified than the other steps, so it is presented in some detail. How the average liquid water content of clouds would change is unknown, so we hold it constant in our analysis.
The liquid water content L(g m-3). the number-density of droplets N(m-3), and the droplet radius r (for a monodispersion) are related by
L= (4/3)πr3ρN
where ρ is the density of water in the appropriate units. Various studies have examined the effect of holding one of these three variables constant while the other two change. Paltridge39, Charlock40, and Somerville and Remer41 considered cloud albedo changes due to a climatic warming which might increase the liquid water content of clouds. They held r fixed so that the increase in cloud albedo was due to increase of N. Bohren42 showed that the increase of albedo would be somewhat less if N was instead held fixed (that is, assuming that CCN concentration did not change), so that the droplets increased in size rather than number.
Table 1: Climatic effect caused by increasing CCN concentration over the ocean
| a Global annual average cloud cover (ocean areas only) | ||
| Cloud type[1] | Ocean area covered (%) | Earth covered by oceanic clouds (%) |
| Non-overlapped St/Sc[2] | 25.2 | 17.6 |
| Non-overlapped As/Ac[3] | 10.8 | 7.5 |
| As/Ac overlapped with St/Sc[4] | 8.8 | 6.1 |
| Nimbostratus. cumulus, cumulonimbus | not applicable (optically thick, high albedo) | |
| Cirrus [5] | not applicable (ice) | |
| Total cover of oceanic stratiform water clouds (As/Ac+St/Sc) not overlapped with cumuliform clouds | 44.8[6] | 31.2 |
| b Example: effect on surface climate due to increasing CCN concentration N by 30% while holding liquid water path fixed | ||
| For area covered by oceanic stratiform water clouds | Averaged over Earth’s surface area | |
| Imposed change in N | +30% | |
| Change in reff | -10% | |
| Change in 0.5-0.7-μm albedo at TOC[7] | +0.02 | |
| Change in 0.5-0.7-μm albedo at TOA[8] | +0.018 | |
| Change in solar albedo at TOA[8] | +0.016 | +0.005 |
| Equivalent change in solar constant[9] | -0.7% | |
| Change in global-average surface temperature[10] | -1.3K | |
[1] Only the areas covered by low stratus and stratocumulus (St/Sc) clouds and middle altostratus and altocumulus (As/Ac) are considered, because the change in albedo is smaller for convectivc clouds and for nimbostratus because they are thicker and may have albedos greater than 0.8.
[2] The zonal average amount (fractional areal coverage) of St/Sc over the oceans varies from 18 at low latitude to 50 at high latitude”‘ and averages 34 for the world ocean, average of all four seasons (ref. 61 and additional data from S.G.W. el ai, in preparation). The daytime (sunlit) amount is close to the diurnal average amount. When St/Sc is observed over the ocean, As/Ac is also present above it about 52 of the time*’2. The amount-when-prescnt of oceanic As/Ac is 50 (S.G.W. et aL, in preparation), so 50 x52 =26 of the St/Sc amount is overlapped bv As/Ac. Non-overlapped St/Sc thus covers 34 x (1.0-0.26) = 25.2 of the ocean area or 17.6 of the Earth’s surface.
[3] As/Ac covers 22.4 of the ocean during the daytime (ref. 61 and S.G.W. el ai. in preparation) but only 10.8 of the ocean is covered by As/Ac which does not overlap St/Sc, cumulus, or cumulonimbus (using a procedure parallel to that used in the St/Sc analysis above).
[4] We assume that this two-layer cloud is still optically thin enough for its albedo to be less than 0.8, that is, in the region of Fig. 1 where Aa is insensitive to a.
[5] A change in albedo of low or middle water-clouds may be muted at TOA when those clouds arc partially hidden by higher ice clouds (cirrus). Here we assume that cirrus is thin enough that the change in planetary albedo due to the water-clouds is the same as if cirrus were absent.
[6] If cumulus is also included, the total areal coverage of clouds whose albedo is sensitive to CCN concentration changes from 44.8 to 56.6.
[7] From Fig. 1.
[8] Because of absorption in the 0.6-μm band of ozone above the cloud, the visible-channel albedo at TOA is smaller than at TOC, bv a factor of about 0.9 (ref. 47). A further factor of about 0.9 is needed to convert visible-channel TOA albedo to solar TOA alhedo4′ “\ because cloud albedo is lower in the near-infrared than in the visible. The same factors apply to Aa.
[9] The global average planetary albedo is now 0.30 (ref. 64). so a change in planetary albedo Aa causes the same change in the amount of solar energy absorbed by the Earth-atmosphere system as would a fractional change in solar constant of (1.0-0.3 )Aa.
[10] From Table 1 of ref. 49.
For our hypothesis, we must instead consider the effect of holding L constant while increasing N. This leads to a decrease in mean radius, which causes an increase in total surface area of droplets in the cloud and thus an increase of cloud albedo. The study of this effect was pioneered by Twomey43. His Figs 12.5 and 12.6 show the albedo, a, at cloud top at visible wavelengths as a function of cloud thickness for a reference plane-parallel cloud with uniform droplet radius r=8μm (‘fairly clean maritime conditions’), as well as the albedo which resulted if N was multiplied or divided by 8, causing r to decrease or increase respectively by a factor of 2. (Much of Twomey’s subsequent work on this topic examined the competing effects on cloud albedo due to absorption of sunlight by dark aerosol particles and the increased number of droplets. Only the latter effect is considered here, because H2SO4 and its ammonium salts are transparent in the solar spectrum.) However, his calculations apparently assumed an overhead sun (zenith angle Θ0=0°) and a black underlying surface. To study the effects on the global radiation budget we have repeated Twomey’s calculations using the global average zenith angle Θ0=60° (which causes the cloud albedo to increase relative to Θ0=0°) and assuming an ocean surface as the lower boundary, for many different values of N. Figure 1 shows the change in albedo Δa from that of Twomey’s reference cloud, caused by variation of N/NΘ in the range 8 to 1/8, where NΘ, is the number-density of droplets in the reference cloud. Different thicknesses of the reference cloud are represented on the vertical axis by their albedos. The information in Twomey’s Fig. 12.6 is therefore contained in the right and left edges of Fig. 1 here, which also shows the effect of any smaller changes in N. Our results agree with those ofTwomey where they overlap, showing that the change in albedo due to changing N is not sensitive to solar zenith angle if the results are expressed as a function of the reference albedo rather than of the cloud thickness. Figure 1 shows that the cloud albedo is most sensitive to N at a = 0.5 but that Δa is essentially independent of a for 0.3 < a < 0.8, for small changes in N/NΘ. Figure 1 was calculated for effective droplet radius of the reference cloud r0eff = 8 μm, but it is approximately valid also for any value of r0eff in the range 4-500 μm (see Fig. 1 legend).
Real clouds are not homogeneous, and the albedo of a cloud with a horizontally inhomogeneous distribution of droplets is always less than that of a hypothetical homogeneous cloud 44,45 because the albedo-versus-optical-depth function is nonlinear, concave downwards. We therefore compare observed clouds to modelled clouds not by comparing their optical depths but rather by comparing their albedos. (The changes Δa will also be somewhat smaller for an inhomogeneous cloud than for a plane-parallel cloud, but not by much if the albedos of the different cloud elements are all in the range 0.3-0.8, as can be seen from Fig.1.)
The top-of-atmosphere (TOA) albedo (or ‘planetary’ albedo) measured by satellites in the 0.5-0.7 μm wavelength channel over marine stratus and stratocumulus (St/Sc) varies in the range 0.25-0.65 (ref. 46). Channel albedo at TOA is usually smaller than channel albedo at top-of-cloud (TOC) by a factor of about 0.9 (ref. 47 and S.G.W. and W. J. Wiscombe, personal communication), because ozone above the cloud absorbs radiation at 0.6 μm, so most marine St/Sc probably have visible TOC albedos, a, in the middle region of Fig. 1 where Aa is insensitive to a for small relative changes of N.
Table 1 illustrates the use of Fig. 1. Stratiform water clouds that probably have visible TOC albedos in the range 0.3-0.8 cover about 45% of the ocean (Table 1a). As an example (Table 1b), we increase N by 30% over the ocean only (because DMS cannot compete with anthropogenic sulphur over land), which causes reff to decrease by 10%. From Fig. 1, this causes visible-channel albedo at TOC to increase by 0.02, which causes an increase of 0.016 in planetary albedo averaged over the solar spectrum above these marine clouds, or 0.005 averaged over the entire Earth. (This increase in whole-Earth albedo is smaller than the value 0.006 shown in Fig. 6b of Twomey et al.48 for this example, because we assume the changes in planetary albedo to apply only to the ocean areas.)
If none of the climatic feedbacks causes cloud albedo to change, the increase in planetary albedo of 0.005 is equivalent to a decrease of the solar constant by 0.7% in a climate model, which causes a decrease of 1.3 K in global mean surface temperature TS, when water-vapour and snow-albedo feedbacks (both positive) are accounted for (Table 1 of ref. 49). This reduction in TS caused by reducing the effective radius of cloud droplets by 10% everywhere over the world ocean is about one-third as large as the increase in TSpredicted for a doubling of atmospheric CO2 (ref. 1).
Of course we do not know the relationship between a change in aerosol concentration over the ocean and the resulting change in cloud-droplet concentration N. Theory and experiments (equations 9-1 and 13-41 of ref. 23) indicate that N is proportional to [CCN]P, with p=~0.8. Setting p= 1 for simplicity, we can show that the cloud-mediated effect on planetary albedo discussed above is much larger than the direct radiative effect of non-nucleated aerosol. Using a mass extinction coefficient of 10m2g-1 for NSS-SO42- (ref. 50), a column-mass of 10-3gm-2, and a ratio of backscattering to total scattering of 0.2, the average total backscattering optical depth, δdsp, of aerosol particles is 3 x 10-3, smaller than the Rayleigh backscattering optical depth due to air itself which is ~0.1. This empirically derived quantity is similar to the estimates of Shaw2. A 30% increase in the number of particles (over the oceans only) with no change in their mean size would increase δbsp by 9 x 10-4, increasing the planetary albedo over dark surfaces by the same amount, but having no effect in areas where clouds are present. The average cloud cover over the oceans is 64% (S.G.W. et al., in preparation) so the increase in planetary albedo from aerosol alone is 9 x 10-4 x (1-0.64) = 3.2×10-4 for the ocean or 2.3 x 10-4 for the whole Earth; that is, only about 5% as large as the cloud-mediated increase.
The example we chose for illustration (a 30% change in N) is actually relatively small compared to observed variations. CCN concentrations over the remote ocean can vary with season and time of day, and from one cloud to another, by an order of magnitude or more23; thus even the extreme left and right sides of Fig. 1 may he applicable for some models of climatic change.
This analysis of the climatic effect of changing the CCN population has assumed that the radiative properties of clouds will change only in the solar spectrum, not in the thermal infrared (beyond 4μm wavelength). Changing the size of droplets will not significantly affect the thermal-infrared emissivity of most water clouds, because they are in effect optically semi-infinite and are nearly black bodies at those wavelengths51. Cirrus is the only type of cloud which is normally thin enough for its emissivity to be sensitive to optical thickness, but the ice-particle sizes are unlikely to be affected by variations in the concentration of CCN because ice nuclei are normally much rarer and from different origins than CCN.
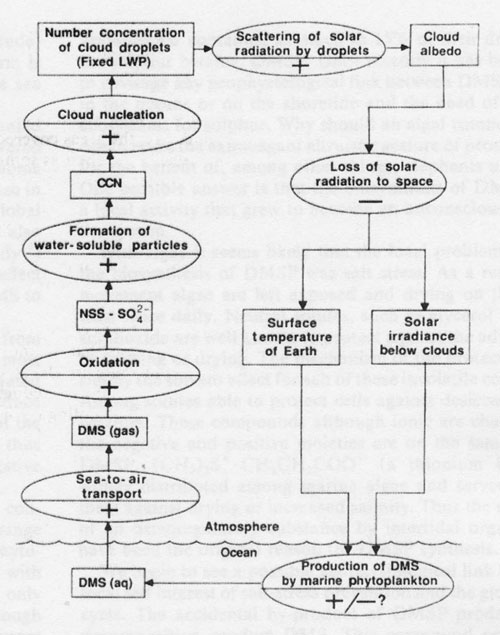
Figure 2. Conceptual diagram of a possible climatic feedback loop. The rectangles are measurable quantities, and the ovals are processes linking the rectangles. The sign (+ or -) in the oval indicates the effect of a positive change of the quantity in the preceding rectangle on that in the succeeding rectangle, following Kellogg’s74 notation. The most uncertain link in the loop is the effect of cloud albedo on DMS emission; its sign would have to be positive in order to regulate the climate.
Global climate and DMS emission
Although we do believe that increased DMS emission should influence both the mass concentration and number population of CCN composed of NSS-SO42- (and hence cloud albedo), we do not understand quantitatively the relationship of source strength of DMS to CCN number concentration. It seems likely that increased DMS fluxes would increase the CCN population, based for example on the observation in polluted air that gas-to- particle conversion produces new particles52. When looking for feedbacks which link the sea-to-air mass flux F of DMS to global climate (Fig. 2), we can consider the variables which make up the flux equation:
F=A · k · Δ c
We can change either the total ocean surface area (A) available for gas exchange, the transfer velocity (k), or the concentration gradient across the air/sea interface (Δc). Because the ocean is highly oversaturated (by at least three orders of magnitude) relative to the atmosphere, the concentration gradient term is essentially identical to the DMS concentration in surface sea water.
A 10% decrease in ice-free ocean-surface area accompanied the last glacial maximum53. During an ice age, the areas covered by ice on the continents and oceans increase, and there is some exposure of continental shelf now under water. The decrease in ocean area during an ice age could lead to a drop in the global flux of DMS to the atmosphere. Climatic changes would also affect the wind field over the ocean (perhaps only slightly54) and thereby the transfer velocity k. The magnitude of this effect on CCN would depend on the relative rates of loss of DMS to the atmosphere and in the water column.
Empirically, we find that the largest flux of DMS comes from the tropical and equatorial oceans5. This suggests that the most important climatic role of DMS is to contribute to elevated cloud albedo over the warmest ocean regions, and thus to reduce the input of heat into the low-latitude oceans. A cooling of the oceans or a reduction in area of the tropical seas could thus lead to a smaller DMS flux, providing a stabilizing negative feedback.
On the other hand, the sea-to-air flux of DMS and consequently the albedo of marine clouds could significantly change as a result of ecological changes which would favour phytoplankton species with large DMS output rates over those with low output rates, or vice versa. At this time we have only examined a few species for their DMS emission rates. Although we know that there are large interspecific differences, we cannot yet relate them to phytoplankton taxonomy. In particular, we are not yet able to use the data on phytoplankton speciation during the geological past, which has been obtained by the CLIMAP program53, to predict the DMS flux during periods of glaciation. As a result, we are left with an incomplete story: we are convinced that the emission of DMS into the marine atmosphere plays a crucial climatic role, but we cannot yet define precisely the processes which regulate the rate of DMS emission.
Geophysiology and homoeostasis
Gaia theory3 suggests that in order to maintain thermostasis on Earth, CO2 is continuously and increasingly pumped from the atmosphere. There is a constant input from tectonic processes, and the long-term sink is the burial of carbonate rock in the sediments. The sink for CO2 is almost wholly biologically determined; without life, CO2 would rise to an abundance of well over 1% by volume. Lovelock and Whitfield55 observed that if climate regulation does take place by pumping of CO2 then the mechanism is now close to the limit of its capacity to operate. Atmospheric CO2 has been reduced from about 30% of the atmosphere at the start of life to the present 300 p.p.m.v., a factor of 1,000. It was suggested that the decrease of CO2 through its declining greenhouse effect had compensated for the monotonic increase of solar luminosity and so the climate had remained constant and suited for life. It cannot be much more reduced without seriously impairing the growth of mainstream plants whose limit is near 150 p.p.m.v.; not much less than the minimum, 180 p.p.m.v., of the last glaciation78.
DMS emission, through its effect on the planetary albedo, shares with C0;-pumping a cooling tendency. So if the DMS production increases with temperature and/or solar irradiance, the sign of its climatic effect would be in the right direction to offset what seems, for the biota, to be an excessive solar flux.
How could the local activity of species living in the ocean evolve to serve in the altruism of planetary regulation? The biogeochemical cycles of carbon and of sulphur are intimately linked and appear to be connected with the regulation of redox potential in both oxic and anoxic ecosystems56,57. The first indication of a geophysiological role for sulphur came from the observations of Challenger8 that marine algae emitted DMS. He found that some species of shoreline algae of the order Polysiphonia contained as much as 15% of their dry weight as the sulphur betaine, DMSP. Until recently it has been difficult to envisage any geophysiological link between DMS production in the oceans or on the shoreline and the need of land-based ecosystems for sulphur. Why should an algal community of the ocean make the extravagant altruistic gesture of producing DMS for the benefit of, among other things, elephants and giraffes? One possible answer is that the biosynthesis of DMS began as a local activity that grew to become an unconscious benefit for the system.
With algae it seems likely that the local problem that led to the biosynthesis of DMSP was salt stress. As a result of tidal movement algae are left exposed and drying on the beach at least twice daily. Neutral solutes, such as glycerol or dimethyl sulphoxide are well known to protect against the adverse effects of freezing or drying. The mechanism of the protective effect is simply the solvent effect for salt of these involatile compounds59. Among solutes able to protect cells against desiccation are the bctaines. These compounds although ionic are charge-neutral; the negative and positive moieties are on the same molecule. DMSP, (CH3)2S+ · CH2CH2COO– (a thionium betaine), is widely distributed among marine algae and serves to protect them against drying or increased salinity. Thus the requirement of an osmoregulatory substance by intertidal organisms may have been the original reason for DMSP synthesis.
We begin to see a possible geophysiological link between the local self interest of salt-stress prevention and the global sulphur cycle. The accidental by-product of DMSP production is its decomposition product DMS. This compound or its aerosol oxidation products will move inland from the shore and deposit sulphur over the land surface downwind of the ocean. The land tends to be depleted of sulphur and the supply of this nutrient clement from the ocean would increase productivity and the rate of weathering and so lead to a return flow of nutrients to the ocean ecosystems. What seems a naive altruism is in fact an unconscious self-interest. Sulphur from DMS can travel farther than the sea-salt aerosol because several steps are involved in the conversion of gaseous DMS to aerosol sulphate; also the resulting aerosol particles are smaller and so have much longer lifetimes.
A large proportion of the current biosynthesis of DMSP is in the open oceans distant from the land surfaces. Is this DMSP also made for the relief of salt stress, or is it a redundant mechanism kept in action because of glacial epochs when the sea or part of it was saltier? Interglacials have occupied only one tenth of the time during the current series of glaciations; this may be too short a period for the devolution of DMSP biosynthesis. Alternatively, it may be that production of DMSP in the open ocean has a different geophysiological basis from that in the continental shelf regions and one that is unconnected with salinity as such.
We have seen how the local self-interest of shoreline algae could lead to the mutual sharing of sulphur and nutrients with land-based organisms. The evolution of a link between ocean climate and DMS production could have happened in a similar way. Ocean organisms are often deficient in nitrogen and to some extent vulnerable to solar ultra-violet. Cloud formation with rainfall would return nitrogen to the ocean and also serve as a sunshade. If either or both of these effects were significant for the health of phytoplankton then species that emitted DMS might be at an advantage.
Is the sulphur cycle also involved in global climate control? Evapotranspiration is known to modify the climate of forests in the humid tropics. The additional cloud cover that comes from the vast water vapour flux of the trees increases the planetary albedo and further cools the surface. The emission of DMS from the oceans seems to act similarly. Through its aerosol oxidation products it alters the properties of clouds, increasing the albedo of the oceanic regions and hence of the greater part of the planet. The link between the biota and climate in both of these processes of cloud formation could be a mechanism for climate control, the clouds serving as do white daisies in the ‘Daisy world’ model of Gaian climate regulation60.
Lastly, DMS is the principal component of the present biogeochemical flux of sulphur to the atmosphere. But it is not the only one; some sulphur is emitted as H2S, COS and CS2. COS, both from direct emission and as an oxidation product of CS2, is stable in the troposphere long enough to be an important source of sulphur to the stratosphere. COS in the stratosphere would be oxidized and produce a sulphate aerosol there. Such stratospheric aerosols scatter sunlight back to space and lead to a cooler climate. The biological variation of COS output is therefore another possible geophysiological means of regulating climate.
Future research needs
We propose that sulphate aerosols derived from the sulphur gases produced by the marine biota arc important determinants of cloud albedo and, as a consequence, the climate. It also seems likely that the rate of DMS emission from the oceans is affected by the climate, thus closing a feedback loop.
There are significant gaps in our knowledge of this proposed feedback system. Most importantly, we need to understand the climatic factors affecting DMS emission. Because some species produce much more DMS than others, we must include the necessary understanding of controls on phytoplankton species abundance. We also need to understand the relationship between DMS concentration in the air and the CCN population, through the intervening aerosol physical processes. Knowing how the area of cloud cover is influenced by CCN is also important.
Nonetheless, the role of the CCN population in controlling albedo, the production of CCN in marine air by the oxidation of DMS from the biota and the sensitivity of the Earth’s temperture to the CCN population seem to be established. Although we do not understand the details of the climatic feedback, it seems that CCN from biogenic DMS currently act to cool the Earth. It is possible that the Earth’s climate has been mediated in the past (for instance, that this feedback has helped to counteract the increasing luminosity of the Sun and / or that it has already counteracted the influence of the recent increase in CO2, and other ‘greenhouse’ gases). However, the data required to demonstrate the latter effect have not been and are not now being acquired.
Acknowledgements
The portion of this work done in the USA was supported in part by NSF grants ATM-82-15337, ATM-83-18028, OCE-83-15733 and ATM-84-07137. The computations were done at the National Center for Atmospheric Research. We thank Marcia Baker, Keith Bigg, Robert Chatfield, lan Galbally, Dean Hegg, Ann Henderson-Sellers, Kendal McGuflRe, Henning Rodhe and Starley Thompson for commenting on an early draft, and Antony Clarke for discussions.
References
To follow.